Single-cell integration benchmark scib


![]()
![]()
Benchmarking atlas-level data integration in single-cell genomics
This repository contains the code for the scib package used in our benchmarking study for data integration tools.
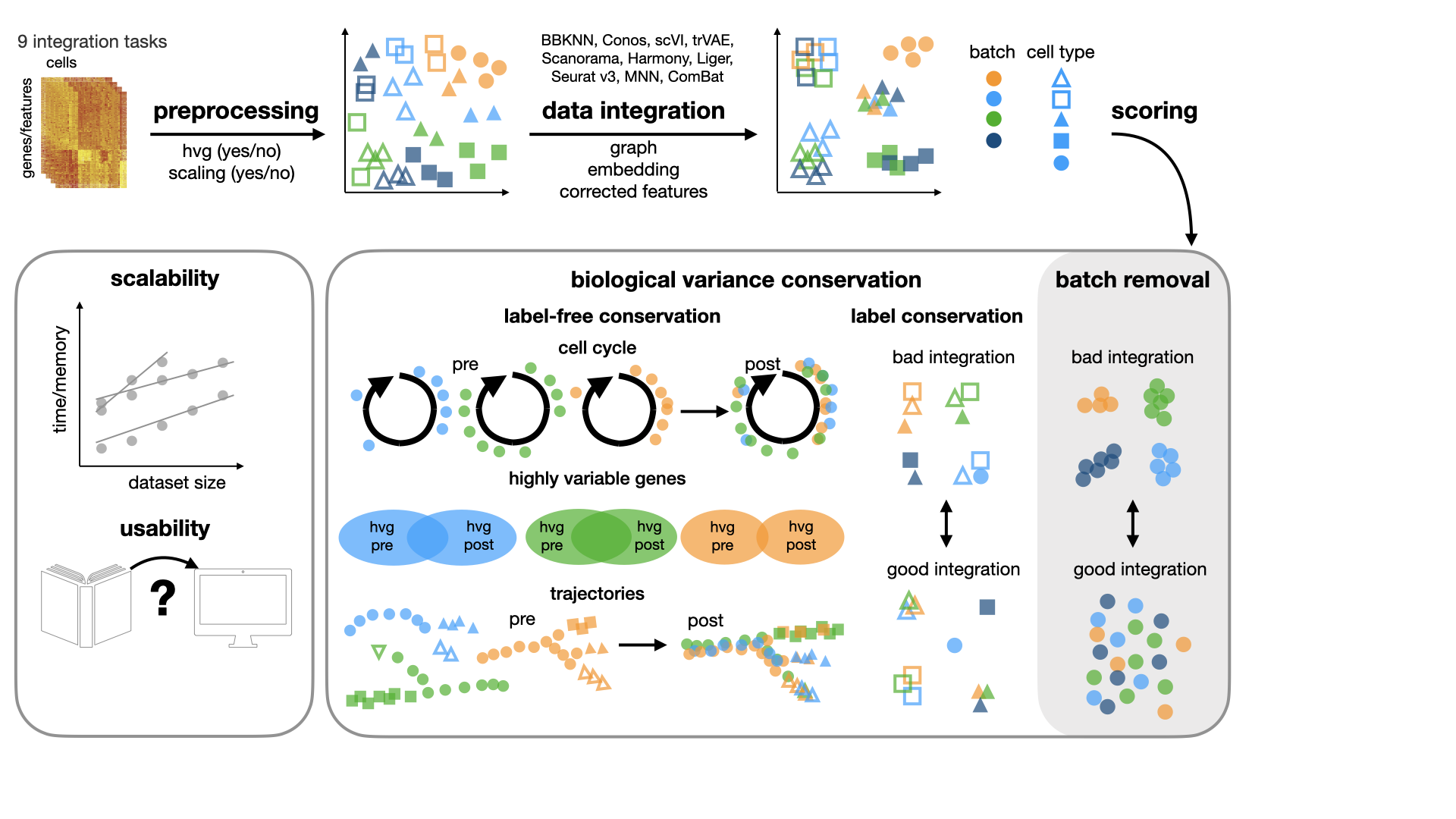
In our study, we benchmark 16 methods (see Tools) with 4 combinations of
preprocessing steps leading to 68 methods combinations on 85 batches of gene expression and chromatin accessibility
data.
Resources
The git repository of the
scibpackage and its documentation.The reusable pipeline we used in the study can be found in the separate scib pipeline repository. It is reproducible and automates the computation of preprocesssing combinations, integration methods and benchmarking metrics.
On our website we visualise the results of the study.
For reproducibility and visualisation we have a dedicated repository: scib-reproducibility.
Please cite:
Luecken, M.D., Büttner, M., Chaichoompu, K. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods 19, 41–50 (2022). https://doi.org/10.1038/s41592-021-01336-8
Package: scib
We created the python package called scib that uses scanpy to streamline the integration of single-cell datasets and
evaluate the results. The package contains several modules for preprocessing an anndata object, running integration
methods and evaluating the resulting using a number of metrics. For preprocessing, scib.preprocessing (or scib.pp)
contains functions for normalising, scaling or batch-aware selection of highly variable genes. Functions for the
integration methods are in scib.integration or for short scib.ig and metrics are under
scib.metrics (or scib.me).
The scib python package is available on PyPI and can be installed through
pip install scib
Import scib in python:
import scib
Optional Dependencies
The package contains optional dependencies that need to be installed manually if needed.
These include R dependencies (rpy2, anndata2ri) which require an installation of R integration method packages.
All optional dependencies are listed under setup.cfg under [options.extras_require] and can be installed through pip.
e.g. for installing rpy2 and bbknn dependencies:
pip install 'scib[rpy2,bbknn]'
Optional dependencies outside of python need to be installed separately. For instance, in order to run kBET, install it via the following command in R:
install.packages('remotes')
remotes::install_github('theislab/kBET')
Metrics
We implemented different metrics for evaluating batch correction and biological conservation in the scib.metrics
module.
Biological Conservation |
Batch Correction |
|---|---|
|
|
For a detailed description of the metrics implemented in this package, please see our publication and the package documentation.
Integration Tools
Tools that are compared include:
Development
For developing this package, please make sure to install additional dependencies so that you can use pytest and
pre-commit.
pip install -e '.[test,dev]'
Please refer to the setup.cfg for more optional dependencies.
Install pre-commit to the repository for running it automatically every time you commit in git.
pre-commit install